Our instrument is an Agilent 8900 ICP-MS. It is capable of analyzing most elements in the periodic table in any sample that can be put into aqueous solution, or ablated with a laser. Detection limits and analytical precision vary widely with analytical protocol, blank contamination, interferences, matrix composition, and isotope abundance, but ranges from <0.1 ppt (parts per trillion) to >100 ppb (parts per billion) in aqueous solution. Analytical relative deviation is typically <3%, 1σ.
Brief notes on sample preparation
- If you don’t know what you are doing, first discuss sample prep with Union Geosciences staff.
- Liquid samples must be in aqueous solution, preferably ~1% HNO3, with or without HCl. Higher concentrations and other acids can be tolerated, but high HF concentrations must be avoided.
- Total dissolved solids in the analyzed solution should generally be <0.1% (1000 ppm), though the autodilution system can permit much higher concentrations if necessary.
- For best results, element concentrations in solution should generally be >0.01 ppb. Having element concentrations <20 ppb is also desirable as it reduces wear on the detector.
- Well-characterized or carefully made standards should be used.
Choosing internal standards
- An internal standard element should not have an isotope that is the same mass as an analyzed isotope.
- Avoid internal standard elements having isotope masses 16 less than an analyzed isotope to avoid MO+ oxide interferences, and elements having an isotope mass exactly twice that of an analyzed isotope to avoid M2+ interferences.
- The internal standard element naturally present in your samples should be overwhelmed by the amount of internal standard you add, so pick elements that are likely to have low abundance in your samples.
- The internal standard isotopes used should have low background, high backgrounds cause more signal variability.
- The internal standards isotopes should be close as possible to the masses of the analyzed elements.
- Internal standard elements should have ionization efficiencies similar to the analyzed elements. This spreadsheet shows ionization efficiencies.
- When using mass-shift gas modes, you should usually use an internal standard element that behaves similarly to the elements you are analyzing in that mode. For example, if you are measuring an element in oxygen gas mode, shifting the analyzed mass up 16 mass units, your internal standard should also be measured as its oxide. You can check element behavior in different gas modes on this spreadsheet.
- Compromises usually have to be made.
Internal standards typically used are:
- Water, no-gas, on-mass gas modes: Sc, Ga, Y, In, Pr, Re, Bi, Tl, Th.
- Water, oxygen mass shift mode: Y, Ru, Ir.
- Water, ammonia mass shift mode: Ge, Y, La, Hf.
- Rocks, no-gas, on-mass gas modes: Ru, In, Re, Tl, Bi.
- Rocks, oxygen mass shift mode: Ru, Ir.
- Rocks, ammonia mass shift mode: Ge.
Containers
If possible, all bottles, volumetric flasks, pipettes, and other things that hold the liquids should be plastic, not glass. Solutions should never be stored in glass.
Digestion vessels
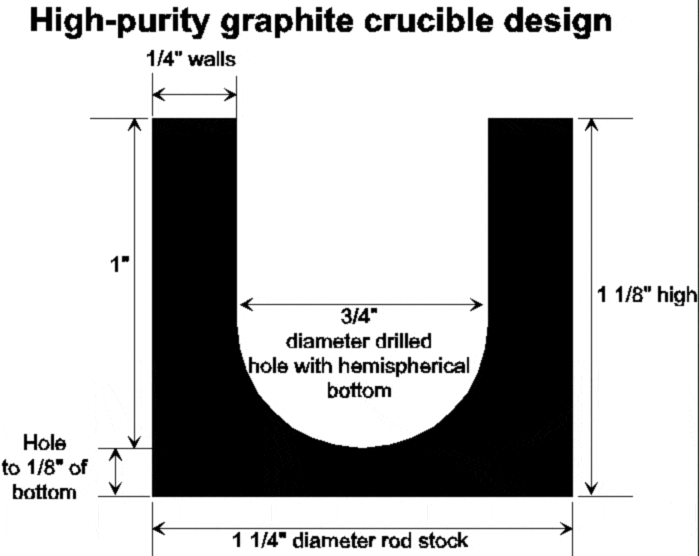
We have used a wide variety of digestion containers, including graphite crucibles for LiBO2 fusions (note: not permitted with our present instrument), 17 ml and smaller Teflon Savillex containers for low-pressure acid digestions, cheap polypropylene or polystyrene test tubes for easily dissolved samples like calcite or phosphate shells, and 30 ml Teflon Picotrace high-pressure digestion vessels.
Reagents
Reagents should preferably be of “ultra high-purity” grade, not the commonly used “reagent grade”. 18 MΩ deionized or sub-boiling distilled water should also be used.
Acids
HNO3 is preferred because it generally causes the fewest problems with polyatomic ion interferences. HCl, HClO4, and H2SO4 result in in a other polyatomic ion interferences. Many of these interferences can be enormously reduced or made irrelevant with proper use of the reaction cell gasses, and mass shifting. HF can be used so long as it is largely removed prior to analysis so it doesn’t damage the glassware, or if the HF-resistant sample introduction system is installed.
Cleanliness
Although clean room facilities are not strictly necessary for preparation of routine samples and analysis of routine elements, it is extremely important to keep the work area clean, especially free of dust, including dust falling from above. Don’t assume new plasticware is free of contamination, clean or test it. If necessary, Teflon digestion vessels should be cleaned under the same conditions under which sample dissolution is done.
Standards
Use standards as similar as possible to the unknowns. Because of the linear response of the instrument over 9 or more orders of magnitude (ppq-ppm), use of a blank and one well-characterized standard is possible, though perhaps not wise. It is also wise not to use standards having element concentrations vastly different than the unknowns. Here is a list of solid standards we have.
Timeliness of sample preparation
Most elements, including the rare earths, seem to be stable in dilute acid solutions forever. Other elements are not stable, depending on the host solution composition. For example, elements such as Ti, Nb, and Ta tend not to be stable in most solutions, though a little fluoride can stabilize these. Time also allows contaminants in the storage containers to leach into solution, or elements in solution to adsorb to the container walls. In general, it is better to analyze samples quickly rather than waiting a long time.
Sticky elements
Certain elements are “sticky” in the ICP-MS sample introduction system. The offenders I have come across can include Li, B, platinum group elements, Ag, Au, Hg, Nb, Ta, and W. Some can be rinsed from the sample introduction system by extended (>1 minute) wash times between samples with ≥5% HNO3. Others can be washed out by adding different acids, such as HCl, if the element complexes well with chloride (e.g., many transition elements, “soft” cations). Sticky sulfide-forming elements like Ag, Au, Hg can be washed out with an L-cysteine solution.
This ICP-MS was funded National Science Foundation grant MRI-1726075.

{kind=link}